


The awarding of the 2024 Nobel Prize to AlphaFold2 marks an important moment of recognition for artificial intelligence’s role in biology. What comes next after protein folding? This article explores how innovative techniques are revolutionizing biomolecule creation through protein generation, accelerating drug discovery and materials science.
From Structure Prediction to Real-World Drug Design
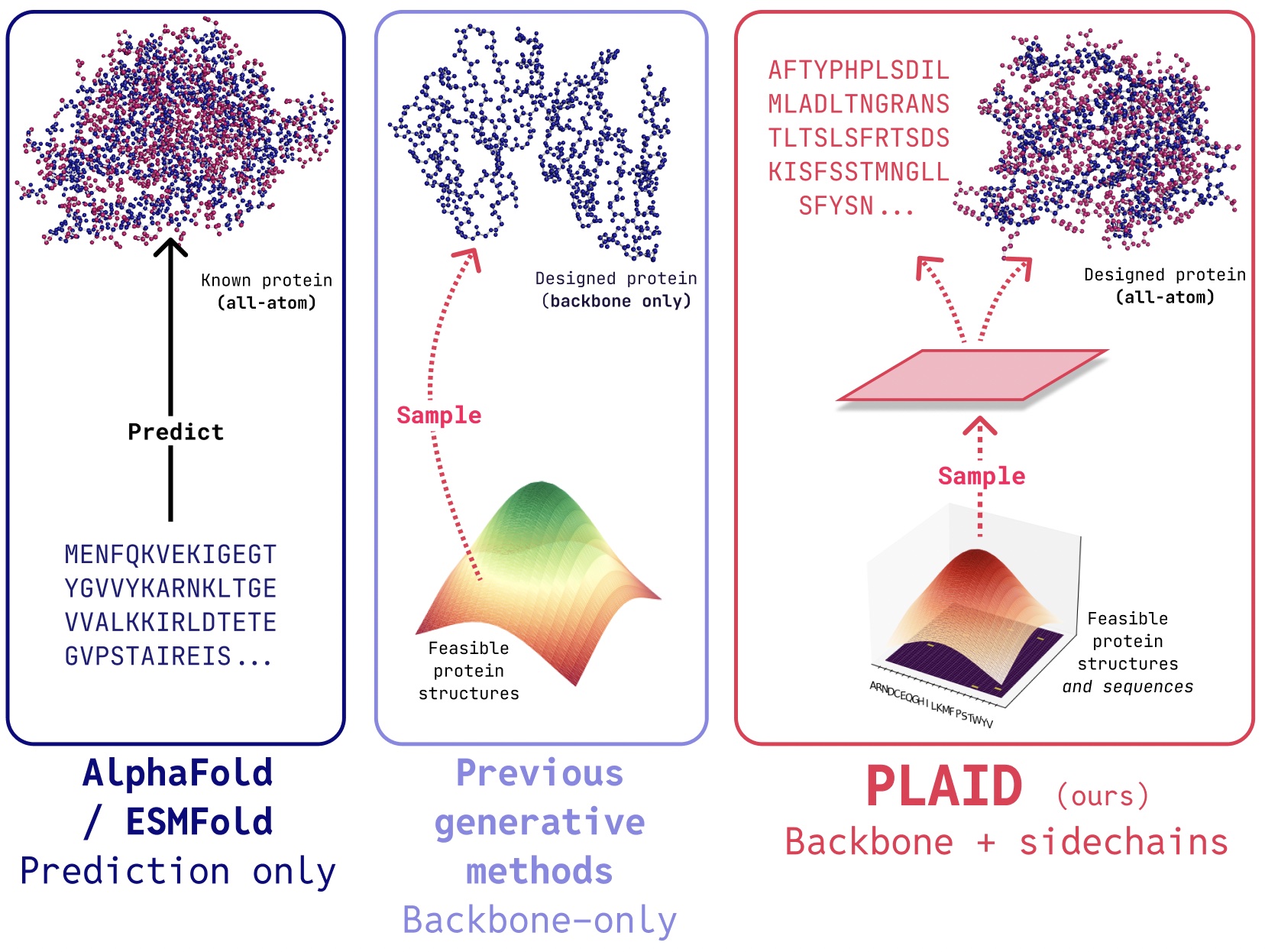
While recent works demonstrate promise for the ability of diffusion models to generate proteins, there still exist limitations that make them impractical for real-world applications. Firstly, many existing generative models only produce backbone atoms. To produce an all-atom structure and place sidechain atoms, a sequence is needed, creating a multimodal generation problem requiring simultaneous discrete and continuous modality generation. Secondly, proteins intended for human use need to be humanized to avoid immune system destruction. Furthermore, drug discovery involves complex constraints; for example, even after addressing biological factors, solubility might dictate tablet formulation over vials.
Generating “Useful” Proteins
Simply generating proteins isn’t as valuable as controlling generation to obtain useful proteins. Let’s consider how we’d control image generation via compositional textual prompts, which mirrors the approach used in protein generation.

The ultimate goal is to control generation entirely via a textual interface, but here we consider compositional constraints for function and organism axes as a proof-of-concept. This allows for greater control in the process of protein generation.

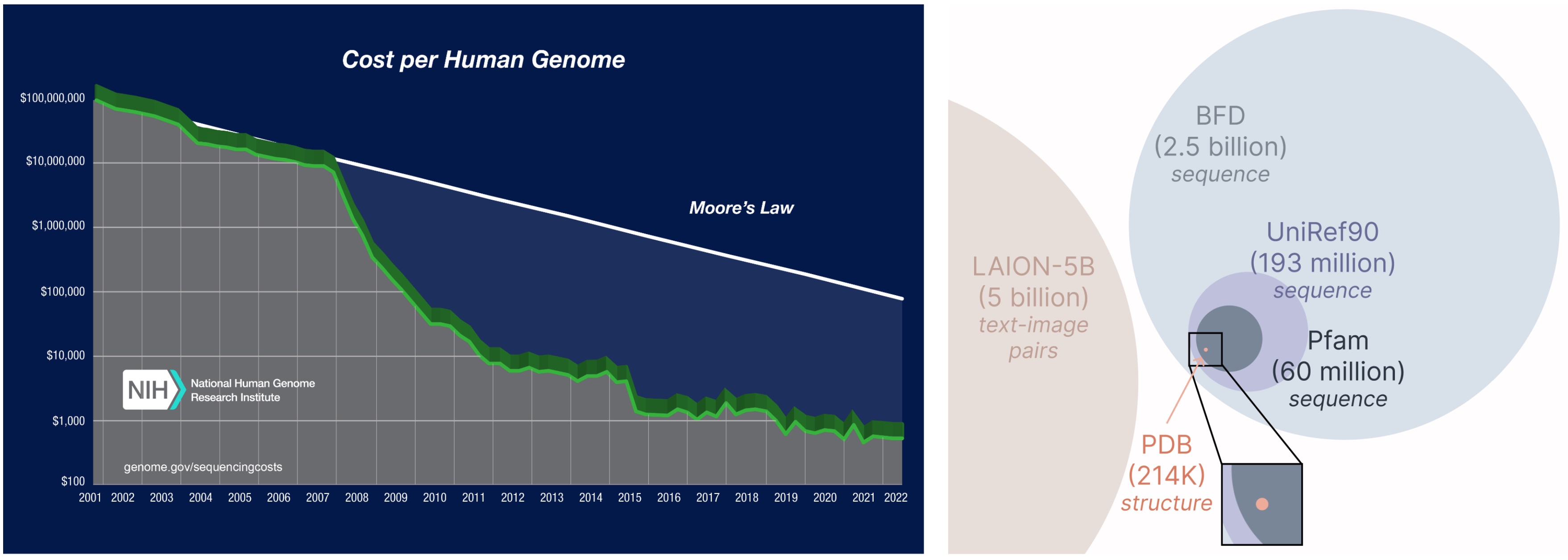
Training Using Sequence-Only Training Data
Notably, this method only requires sequences to train the generative model! Generative models learn the data distribution defined by its training data, and sequence databases are considerably larger than structural ones. Therefore, utilizing these abundant datasets is crucial for effective protein generation.
How Does It Work?
We can train the generative model to generate structure using only sequence data by learning a diffusion model over the latent space of a protein folding model. During inference, we decode structure from frozen weights of the protein folding model. We use ESMFold, a successor to AlphaFold2.
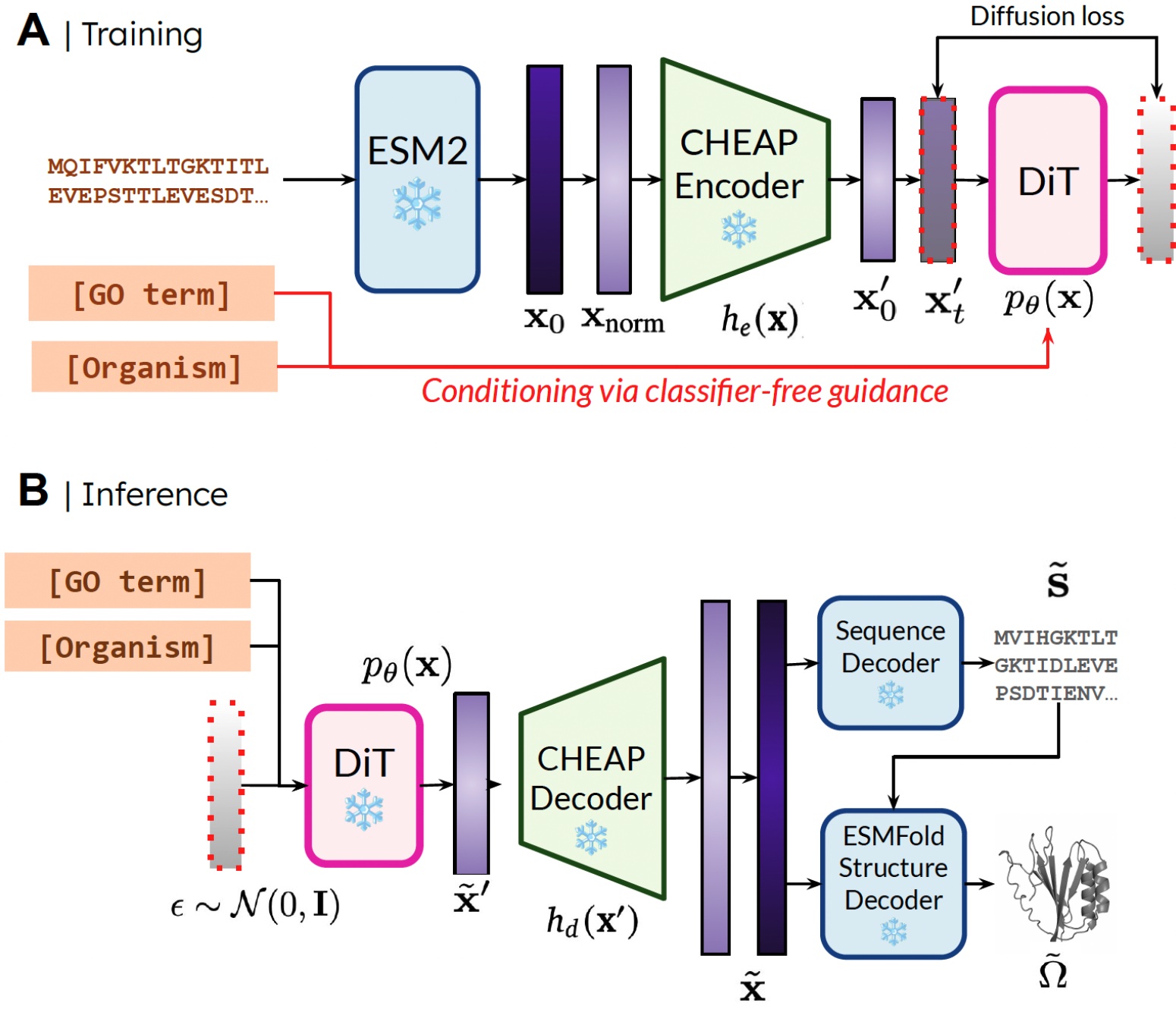
Compressing the Latent Space of Protein Folding Models
The latent space of ESMFold requires a lot of regularization. To address this, we propose CHEAP (Compressed Hourglass Embedding Adaptations of Proteins), where we learn a compression model for the joint embedding of protein sequence and structure. In addition to improving efficiency, this approach enhances the capabilities of protein generation.
What’s Next?
This method can be adapted to perform multi-modal generation for any modalities where there is a predictor from an abundant modality to a less abundant one. If you are interested in collaborating, reach out!
Further Links
@article{lu2024generating,
title={Generating All-Atom Protein Structure from Sequence-Only Training Data},
author={Lu, Amy X and Yan, Wilson and Robinson, Sarah A and Yang, Kevin K and Gligorijevic, Vladimir and Cho, Kyunghyun and Bonneau, Richard and Abbeel, Pieter and Frey, Nathan},
journal={bioRxiv},
pages={2024--12},
year={2024},
publisher={Cold Spring Harbor Laboratory}
}
@article{lu2024tokenized,
title={Tokenized and Continuous Embedding Compressions of Protein Sequence and Structure},
author={Lu, Amy X and Yan, Wilson and Yang, Kevin K and Gligorijevic, Vladimir and Cho, Kyunghyun and Abbeel, Pieter and Bonneau, Richard and Frey, Nathan},
journal={bioRxiv},
pages={2024--08},
year={2024},
publisher={Cold Spring Harbor Laboratory}
}
Check out our preprints (PLAID, CHEAP) and codebases (PLAID, CHEAP).
Source: Read the original article here.
Discover more tech insights on ByteTrending.
Discover more from ByteTrending
Subscribe to get the latest posts sent to your email.









